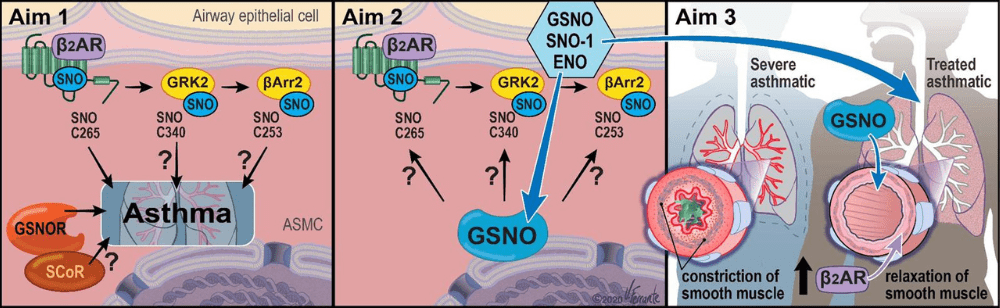
Asthma afflicts over 7% of Americans and is the result of chronic inflammation of the airways leading to airway remodeling and hyperresponsiveness that impedes airflow. Severe asthma is asthma that remains problematic despite maximal intervention with conventional asthma therapies, and it accounts for the majority of the mortality and cost of asthma. Dysfunction of airway β2-adrenergic receptor (β2AR) signaling contributes to severe asthma pathogenesis, but the precise signaling mechanisms responsible are unclear. While activation of β2AR using inhaled “β-agonist” drugs is a mainstay of acute asthma treatment, overactivation of β2AR is detrimental and can be fatal. Airway β2ARs signal both through heterotrimeric G proteins and through G protein-coupled receptor kinase (GRK)/β-arrestin pathways that also mediate receptor phosphorylation, desensitization, and internalization.
Through a long-standing collaboration, the Stamler and Gaston groups have found that airways are regulated by nitric oxide (NO) through S-nitrosylation of thiols to form S-nitrosothiol (SNO), including on cysteine residues in proteins, a post-translational modification that alters protein functions. In addition, SNO forms on low molecular weight thiols, including glutathione to form SNO-glutathione (GSNO). We demonstrated that inhaled GSNO elevates lung protein-SNO and is protective in asthma, identified the enzyme SNOglutathione reductase (GSNOR) that inactivates GSNO, and demonstrated that mice lacking GSNOR are protected from developing asthma. Since the β2AR can activate NO synthase in the airways to generate NO, there is a need to discover how this promotes endogenous SNO-mediated bronchoprotection. Our recent work has shown that β2AR is S-nitrosylated after activation, and preventing SNO-β2AR with a point mutation augments β2AR signaling. Importantly, mice bearing β2AR with a knock-in of this mutation are protected from developing asthma. We have previously shown that β2AR regulators GRK2 and β-arrestin2 are S-nitrosylated to inhibit their activity to desensitize the β2AR, and mice bearing GRK2-C340S and β-arrestin2-C253S knock-in exhibit heightened β2AR activity and worsened injury in cardiac models. We have begun to test the efficacy of inhaled GSNO to affect bronchorelaxation and improved lung function in patients, and these data and clinical samples uniquely position us to examine the role of S-nitrosylation in regulating the β2AR pathway from bench to bedside.
The Central Hypothesis of Project 1 is that the β2AR signaling system is a key target and mediator of SNO-GSNOGSNOR protective effects in the airways in severe asthma. Our studies will define the role of S-nitrosylation of specific β2AR signaling pathway components in a murine model of asthma, delineate the roles of inhaled GSNO and of GSNO dinitrosylases on β2AR signaling components in murine models of asthma and in human lung primary cells, and demonstrate that inhaled GSNO improves both airway flow and β2-agonist responsiveness in severe asthma. This work complements the clinical aims of Project 2 and Project 3 by providing a mechanistic link between NO/GSNO/GSNOR actions and the regulation of β2AR signaling in severe asthma.